医疗器械CE认证指令为93/42/EEC(MDD),如果为带电的医疗器械,还会涉及低电压指令2006/95/EC(LVD)指令和电磁兼容指令2004/108/EC(EMC)指令。
相关的电气测试也需送检测试,常用的欧盟标准有:EN60601-1医用电气设备第一部分:安全通用要求;EN60601-1-1;EN60601-1-2
欧盟委员会针对医疗产品制定了三个指令:
1. 有源植入性医疗器械指令(AIMD, 90/335/EEC),适用于心脏起搏器,可植入的胰岛素泵等有源植入性医疗器械。AIMD于1993年1月1日生效。过渡截止期为1994年12月31日,从1995年1月1日强制实施。
2. 活体外诊断器械指令(IVD)90/385/EEC,适用于血细胞计数器,妊娠检测装置等活体外诊断用医疗器械。
3. 医疗器械指令(Medical devices)93/42/EEC,适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械(敷 料、一次性使用产品、接触镜、血袋、导管等);以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。该指令有欧盟委员会在1993年6月14日颁布,并于1998年6月14日起强制执行。
目录
1.医疗器械产品2014年7月正式纳入RoHS 2.0 管控范围
2011年7月1日欧盟在其官方公报上正式发布RoHS修订指令2011/65/EU(RoHS 2.0),该指令于2013年1月3日全面实施。
RoHS 2.0 纳入CE标志要求,成为欧盟CE标志指令之一。电子电气产品只有在符合低电压指令(LVD)、电磁兼容(EMC)、能源相关产品(ErP)和RoHS 2.0的指令要求,才能粘贴CE标志,出具符合性声明,而且RoHS 2.0还同时要求制造商出具支撑性技术文档,并保留十年。管控产品范围扩大,新增第 8 类医疗设备、第 9 类监控设备和增加第 11 类其他电子电气设备。
医疗器械产品RoHS 2.0实施日程表
2014年7月22日, 医疗器材纳入RoHS 2.0 管控范围2016年7月22日,体外诊断医疗设备月纳入RoHS 2.0 管控范围主动植入式医疗设备(暂时豁免)
2.欧盟医疗器械定义及分类:
医疗器械指单独或者组合使用与人体的仪器、设备、器具、材料或其他物品,包括所需要的软件;其适用旨在达到下列预期目的:
1. 对疾病的预防、诊断、监测、治疗或减缓;
2. 对受伤或残障的诊断、监护、治疗、环节或修补;
3. 对解剖或生理过程的研究、替代、调节;
4. 妊娠控制
医疗器械分类:
欧盟把医疗器械产品分为四类:第Ⅰ类、第Ⅱa类、第Ⅱb类、第Ⅲ类
医疗设备供应商必须确保设备制造符合或超过所需的安全和性能标准。 在满足这些会对设备设计、开发、测试、制造、包装和标记造成影响的管理要求之后,产品即可投放市场。
1. 医疗欧盟MDD指令EN标准的测试CE认证
EN/IEC 60601-1(一般安全要求)
EN/IEC 60601-1-xx(附属要求)
EN/IEC 60601-2-xx(特定)系列标准
对医疗设备进行EMC测试
医疗设备指令,Class I-m(测量)、I-s(无菌)、IIa、IIb、III
EC合规性声明;编制试验报告和对技术文件(MDD附件VII)进行所需的文档化。
EC类审核证书(MDD附件III)。
EN ISO 13485全面质量保证(MDD附件II)。
EN ISO 13485产品质量保证(MDD附件V)。
MDD附件VI产品质量保证
MDD附件IV之EC审核。
2. ISO13485管理系统认证
ISO 13485:2003是一种用于医疗设备的质量管理系统。若要符合欧洲管理要求,首先要符合ISO 13485:2003。ISO 13485:2003有助于建立一种通过美国FDA认证所必需的质量管理系统。
在欧洲,在获准出售医疗设备之前,必需按照EEC法令(93/42/EEC)对其合规性进行评估。若要获得CE认证证书和在欧盟销售医疗设备的许可,必须由指定机构根据ISO 13485:2003和93/42/EEC指令对公司质量管理系统进行认证。
3. 环境合规性
4.医疗器械CE认证产品分类
医疗器械产品做CE认证怎么分类的?医疗器械产品分为几类?各类医疗器械产品CE认证怎么做?医疗器械CE认证产品分类规则?
医疗器械指令附录九中详定18条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。
医疗器械产品分类规则:
1、规则应用由器械的预期使用目的决定;
2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3、附件可以和其它一起使用的器械分开单独分类;
4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:
1.时 间: 暂时 (<60分钟)、短期(<30天)、长期(>30天)
2.创伤性:非创伤、通过孔径创伤,外科创伤 、植入。
3.适用位置:中央循环、中枢神经系统,其它地方。
4.能量供应:无源,有源。
规则1~4、所有非创伤性器械均属于I类,除非他们:
用于储存体液(血袋例外) II a类
于Ila类或更高类型的有源医疗器械类 II a类
改变体液成分 II a/II b类
一些伤口敷料 II a/II b类
5. 规则5、侵入人体孔径的医疗器械
暂时使用(牙科压缩材料、检查手套) I类
短期使用(导管、隐形眼镜) II a类
长期使用(正常牙线) II b类
6. 规则6-8、外科创伤性器械
再使用的外科器械(钳子,斧子) I类
暂时或短期使用(缝合针。外科手套) 11a类
长期使用(假关节,眼内晶体) II b类
与中央循环系统(CCS)或中枢神经系统接触的器械 III类
7. 规则9、给予或交换能量的治疗器械 II a类
(肌肉刺激器、电钻、皮肤光疗机、助听器)一一种潜在危险方式工作的 II b类
(婴儿培养箱、高频电刀、超声碎石机、X光机)
8.规则10、诊断器械
提供能量(核磁共振,超声诊断仪) II a类
诊断/监视体内放射药物分布 II a类
(r照相机、正电子发射成像仪)
诊断/监视生理功能(心电图、脑电图) II a类
危险情况下监视生理功能 II b类
(手术中的血气分析仪)
发出电离辐射(X射线诊断议) II b类
9.规则11 控制药物或其他物质进出人体的有源器械 II a类
(吸引设备、供给泵)
如以一种潜在危险方式工作 II b类
(麻醉机、呼吸机、透析机、高压氧舱)
10.规则12.所有其他有源医疗器械属于I类
(观察灯、牙科椅、轮椅、牙科用治疗灯、记录处理观察诊断图象用的有源器械)
11.规则13、与医用物质结合的器械(含杀精子的避孕套、含抗生素的牙髓材料) III类
12.规则14、避孕用具(避孕套、子宫帽 II b类 ) II b/III类 (子宫内避孕器 III类)
13.规则15、清洗或消毒的器械: 医疗器械(内窥镜消毒) II a类、接触镜(消毒液、护理液) II a类
14.规则16、用于记录X射线图象的器械(X光片) II a类
15.规则17、利用动物组织的器械(生物)心脏瓣膜、肠线、胶原) III类
16.规则18、血袋 II b类
5.医疗器械产品CE认证准备工作
医疗器械产品CE认证应该准备什么?要做什么准备工作?医疗器械产品怎样做CE认证?
1. 医疗器械产品要顺利通过欧盟CE认证,需要做好三方面的工作:
第一,收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准。上海曼励您身边的认证专家,专业解答技术法规和标准,为您节省精力和时间。
第二,企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN标准的要求,贯彻到企业产品的设计开发和生产制造的全过程。
第三,企业必须按ISO9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证。针对不同的产品采用的方式不同,具体参看下面的医疗器械产品分类
2. 医疗器械CE认证应遵循的欧盟技术法规和EN标准:
对于目前欧盟已发布的18类工业产品指令,从这些指令的结构看,它们可分为垂直指令和水平指令。垂直指令是以具体产品为对象,如医疗器械指令;水平指令适用于各种产品系列,如电磁兼容性指令,它适用于全部电器及电子零部件产品。
对于医疗器械,适用的指令有第十四项、第一项和第五项,即:93/42/EEC医疗器械指令、低电压指令2006/95/EC(LVD)指令和电磁兼容指令2004/108/EC(EMC)指令。
支持这些指令的欧盟标准是:
(1)EN60601-1医用电气设备第一部分:安全通用要求;
(2)EN60601-1-1医用电气设备第一部分:安全通用要求及第一号修正;
(3)EN60601-2-11医用电气设备第二部分:γ射束治疗设备安全专用要求;
(4)EN60601-1-2医用电气设备第一部分:安全通用要求1.2节并行标准电磁兼容性——要求和测试。其中第(1)、(2)、(3)项标准是伽玛刀低电压(LVD)测试的依据:第(4)项标准是伽玛刀电磁兼容性(EMC)测试的依据。
3. 医疗器械CE认证程序、内容
欧盟把医疗器械产品分为四类,即:第Ⅰ类、第Ⅱa类、第Ⅱb类、第Ⅲ类。
第Ⅰ类产品要加贴CE标志,可采取自行宣告的方式。即厂商编制产品的技术文件档案,同时自行按有关EN标准对产品进行测试或委托有能力的试验室进行测试合格。 第Ⅱa类、第Ⅱb类、第Ⅲ类产品要加贴CE标志,则必须由欧盟指定的验证机构验证。欧盟还规定,这几类产品获得CE认证的先决条件是制造厂需能过ISO9000+ISO13485质量体系认证,取得ISO9000+ISO13485质量体系认证证书,且证书的颁发单位应为欧盟认可的认证机构。ISO9000+ISO13485质量体系认证和CE认证可同时进行,但CE证书必须待ISO9000+ISO13485质量体系认证通过后,方可予以颁发。
6.医疗器械CE认证测试项目
医疗器械CE认证根据产品不同会涉及低电压指令、电磁兼容指令相关测试、生物相容性测试等
具体的对应测试项目有
| 测试类别及依据 | 测试项目 |
|
安规 Safety
IEC/EN/UL 60601-1 |
输入功率Power input |
| 剩余电压Voltage limitation | |
|
防除颤测试 Defibrillation-proof applied parts |
|
|
接地阻抗 Earthing and potential equalization |
|
| 漏电流测试Leakage current test | |
| 耐压测试Dielectric voltage withstand | |
|
外壳机械强度测试 Enclosure mechanical strength |
|
| 稳定性测试Stability and transportability | |
| 温升测试Temperature | |
|
溢流,泼洒,泄漏,清洗,消毒和灭菌 Overflow, spillage, leakage, cleaning, sterilization and disinfection |
|
|
单一故障测试 Abnormal operation and fault conditions |
|
| 电源线拉力和扭力测试Cord anchorages | |
| 电源护套弯折测试Cord bending | |
|
变压器过载和短路测试 Transformer overload and short-circuit tests |
|
|
变压器倍压倍频测试 Transformer dielectric voltage withstand |
|
| 球压测试Ball pressure | |
| 坠落冲击测试Drop impact test | |
| 模压测试Mold stress relief test | |
| 等etc. | |
|
电磁兼容 EMC
IEC/EN 61326-2-6, |
传导干扰Conducted Emission |
| 辐射干扰Radiated Emission | |
| 谐波干扰Harmonic Emission | |
| 电压闪烁Flicker Emmission | |
| 静电放电Electrostatic Discharge (ESD) | |
| 辐射抗扰度Radiated Immunity | |
| 快速脉冲群Electrical Fast Transients (EFT) | |
| 工频磁场Power Frequency Magnetic Feld(PFMF) | |
| 电压跌落和中断Voltage Dips and Interruptions | |
| 浪涌Surge | |
| 射频 RF | |
| 等etc. | |
|
生物相容性 Biocompatibility
ISO/EN 10993 |
热源 Pyrogen |
| 植入Implantation | |
| 基因毒性Genotoxicity | |
| 系统毒性Systemic Toxicity | |
| 血液相容性Hemolysis | |
| 皮肤刺激性Skin Irritation | |
| 皮内刺激性Intracutaneous Irrtation | |
| 急性细胞毒性Acute Cytotoxicity | |
| 极大化过敏性Maximization Sensitization | |
|
兔子眼睛刺激性 The Irratation os Rabbit Eyes |
|
|
可靠性 Reliability
IEC 60068, IEC 60529,IEC 60598, |
高温/低温High / Low Temprature |
|
恒温恒湿 Constant Temprature and Humidity |
|
| 振动Vibration | |
| 盐雾Salt Spray | |
| 温度湿度循环Temprature & Humidity Cycling | |
| 寿命分析 | |
| 高加速寿命试验& 高加速应力筛选 | |
| 臭氧老化 Ozone Aging | |
| 防水防尘Waterproof and Dustproof | |
|
震动&温度&湿度 Vitration & Temprature & Humidity |
|
| 高加速寿命试验 HALT/HASS/HASA | |
|
失效分析与预防 Failure Analysis and Prevention
IPC/ECA-J-STD, |
PCB&PCBA失效分析和评估 PCB & PCBA Analysis and Evaluation |
|
IC可靠性设计分析 Reliability Design and Analysis of IC |
|
|
无损检测 Non-destructive Testing |
|
| 涂镀层Coating and Palte Analysis | |
| 材料热学Material Thermal | |
| 材料硬度Materal Hardness | |
| 扫描电镜能谱分析SEM-EDS |
一、分析器械及特点,确定它是否在欧盟的3个医疗器械指令的范围内;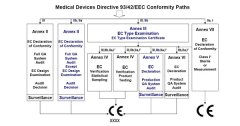
二、确认适用的基本要求;
指令规定,任何医疗器械必须满足相关指令中所规定的预期用途,所以对制造商来说,首先要做的而且是最重要的事情就是确认所有的适用于其产品的基本条件。
三、确认任何有关的欧洲协调标准;
协调标准是由欧洲标淮委员会(CEN)和欧洲电气技术委员会 (CENELEC)制定的公布在欧盟官方杂志上的标准,对于某种医疗器械来说,可能有多种协调标准适用于它,因此在确认哪些协调标准适用于某种产品对应十分仔细。
四、产品分类;
根据指令附录IX的分类规则,医疗器械分成4类,即I、IIa、IIb和III类,不同类型的产品,其获得CE标志的途径(符合性评价程序)不同,因此对制造商来说,如何准确地确定其产品的类型,是十分关键的。
五、确保产品满足基本要求或协调标准的要求并且使证据文件化(技术文档的整理);
制造商应能提出充分的证据(如,由认证机构或其他检测机构依据协调标准进行的检测等)来证明产品符合基本要求。
六、确定相应的符合性评价程序;
如附图I所示,对于IIa、IIb和III类医疗器械的制造商来说,存在着如何选择符合性评价程序途径的问题。主要的区别是选择型式试验的方式,还是选择质量体系的方式,这两种途径各有其特点,制造商应根据自己的实际情况选择最为适合的途径。
七、选择认证机构;
对于IIa、IIb和III类医疗器械,以及无菌的或具有测量功能的I类医疗器械,应选择一个认证机构并进行符合性评价程序。在欧盟官方杂志上公布的认证机构名单上,对每个认证机构可以从事的医疗器械认证范围以及可进行的符合性评价程序途径都有严格的规定,制造商在选择认证机构时,必须非常谨慎,避免造成不必要的损失。
八、起草符合性声明并加贴“CE”认证标志;
可以说符合性声明是最重要的文件。每一种器械必须包括医疗器械指令的附录中所描述的符合性声明。
附录:怎样选择认证机构
认证执构是一个由欧盟认可的审核和认证机构,它可从事医疗器械指令的附录中所描述的一种或多种符合性评价程序。认证机构必须位于欧盟的某个成员国内。
选择认证机构是制造商面临的极其关键的问题之一。为了能够有效地工作应和认证机构建立长期和密切的联系。应对非常慎重地选择“伙伴”所花费的时间和财力应被认为是一项公司未来的投资。
一般未说,制造商在选择认证机构的时候,应考虑下列因素:
⑴ 医疗器械认证方面的经验;
⑵ 所熟悉的医疗器械的范围;
⑶ 拥有的专业特长,如电磁兼容、软件等;
⑷ 与一些委托方的关系及委托方的资格;
⑸ 被授权的医疗器械认证范围;
⑹ 被授权的可进行的符合性评价程序;
⑺ 对已有证书的态度;
⑻ 费用;
⑼ 地点和工作语言;
8.医疗设备CE认证技术档案所需内容:
1、生产商/或欧洲代表名称地址;
2、产品及型号描述;
3、EC符合声明书;-上海曼励指导下完成
4、风险评估;-上海曼励指导下完成
5、基本安全点检表;
6、适用之调合标准/或其他标准;
7、市场反馈及抱怨分析;
8、使用说明及标签;
9、授权代表;
10、线路、图表(适用的话);
11、计算书、测试报告或其它证明材料;
12、检验过程及过程描述;
13、灭菌或其它特殊过程(适用的话);
14、灭菌类产品的包装材料及方法;
15、质量体系、质量手册;
"技术文档"是欧盟医疗器械CE认证中重要的技术文件。它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。各欧盟指令对于"技术档案"的要求有所差别,在这里谨以中国出口企业最常用的“医疗器械”的要求为例,加以说明。
医疗器械指令93/42/EEC要求"技术档案"可能包含下列项目:
A、 企业的质量手册和程序文件;
B、企业简介及欧洲代理名称、联系方式;
C、CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料);
1、产品名称、分类及引用标准条款的简要描述
2、产品概述(包括类型和预期用途
a) 产品的历史沿革
b) 技术性能参数
c) 产品配合使用的附件、配合件和其它设备清单
d) 产品的图示与样品
e) 产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施(EN1441 产品服务危险分析报告)
5、生产质量控制
a) 产品资料和控制文档(包括产品生产工艺流程图)
b) 产品的灭菌方法和确认的描述
c) 灭菌验证
d) 产品质量控制措施
e) 产品稳定性和效期的描述
6、包装和标识
a) 包装材料说明
b) 标签
c) 使用说明书
7、技术评价
a) 产品检验报告及相关文献
b) 技术概要及权威观点
8、潜在风险评价
a) 产品潜在风险测试报告及相关文献
b) 潜在风险的概要及权威观点
9、临床评价
a) 产品临床测试报告及相关文献
b) 临床使用概述及权威观点
1、临床研究(包括:物理性能,生化、药理 、药动及毒性研究,功效测试,灭菌合格证明,药物相容性等);
2、生物兼容性测试(A)EN30993 第一部分要求:细胞毒性、感光性、刺激-皮内反应、急性全身中毒、致热性、亚急性中毒、遗传毒性、植入溶血性;B)支持测试:慢性中毒、致癌性、再生性/生长性毒素、生物动因退化。);
3、临床资料(需要临床研究或描述临床研究);
4、包装合格证明(EN868);
5、标签、使用说明(EN980、EN1041);
6、结论(设计档案资料的接受、利益对应风险的陈述);
上述文件都必须用欧盟官方语言之一(英、德、法文)编写,但使用说明必须用使用者所在国语言编写。所有文件应在最后一次出货后,至少保存五年。